






")



 – कैलकुलेटर और प्रीमियम चार्ट")

")




A person with Creutzfeldt-Jakob disease (CJD) has a rare disease that quickly deteriorates their brain. The symptoms of this disorder resemble dementia as it progresses and affects your brain.
When you have CJD, aberrant proteins called prions accumulate in your brain cells, causing harm and eventual cell death. The illness is quite serious, and its consequences intensify over time. Unfortunately, there is no known cure, treatment, or even way to halt the disease’s progression before it is deadly.
Who is impacted by it?
People between the ages of 50 and 80 are usually affected by Creutzfeldt-Jakob disease, which is more common as one ages. On the other hand, the hereditary subtype of CJD typically manifests sooner, primarily in the 30–50 age range. Overall, it affects men and women equally.
Differential CJD
Variant CJD (VCJD) is a subtype of CJD that affects consumers who consume meat from cattle that have been infected with bovine spongiform encephalopathy (BSE).
What is the prevalence of this condition?
CJD is very rare, with 1 to 2 cases happening in every 1 million people worldwide. About 350 people receive a diagnosis of CJD in the U.S. each year.
What happens when you have Creutzfeldt-Jakob disease?
CJD is a degenerative brain disease, meaning it causes damage to your brain that worsens over time. Experts classify it as a “transmissible spongiform encephalopathy” (TSE). The incubation period, which is the time it takes from when you first get the disease to when it starts causing symptoms, can range from months to years.
One to two cases of CJD occur globally for every million people, making it an extremely rare disease. In the United States, about 350 persons are diagnosed with CJD annually.
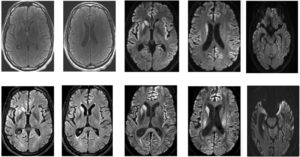
What are the signs and symptoms of Creutzfeldt-Jakob disease?
As a degenerative brain illness, CJD damages your brain gradually and gets worse over time. It is categorized as a “transmissible spongiform encephalopathy” (TSE) by experts. The amount of time between when you initially contract the disease and when symptoms appear can vary greatly, ranging from months to years. This period is known as the incubation period.
Small areas of brain injury, known as clusters, are brought on by TSE disorders. Your brain becomes disfigured by this injury, resembling a sea sponge or a Swiss cheese with plenty of holes and hollows.
You lose the control over the abilities in the damaged area of your brain as this injury develops. In general, a wide range of symptoms are experienced by those with CJD, such as memory loss, cognitive difficulties, uncontrollable muscular spasms or mobility issues, and more. Because of the extent of damage it causes, CJD is ultimately fatal.
Signs and Origins
What signs of the disease Creutzfeldt-Jakob exist?
The following are the most typical symptoms, arranged from early to late in the disease:
- Memory issues and forgetfulness.
- Lost in thought and direction.
- Personality and behavior shifts.
- Issues with vision or the processing and interpretation of what you observe.
- Delusions or hallucinations.
- Issues with motor function (ataxia).
- Issues with balance can impact steadiness whether standing or walking.
- Spasms and uncontrollably moving muscles (dystonia).
- Seizures.
- Immobility.
- Wasting (sometimes referred to as muscle atrophy, which is the fast loss of weight and muscle mass).
Creutzfeldt-Jakob disease: What causes it?
CJD is brought on by prions, which are faulty proteins in the brain. Proteins are chemical molecules that require a certain shape in order to function (much like a lock that unlocks when the correct key is inserted).
When proteins are misshaped, they cannot be used by your cells and cannot be broken down by your body. Those proteins steadily accumulate in the neurons, the brain’s main brain cells, and eventually cause the neurons to break down. For whatever reason, your body can produce defective proteins, and those misshaped proteins can lead to degenerative brain illnesses like Alzheimer’s.
However, prion-based illnesses differ significantly. Prions change normal proteins into more prions rather than causing a steady accumulation of defective proteins. More proteins become prions as the number of prions increases. The rate of conversion increases with the number of prions. That’s why the symptoms of CJD progress so swiftly from moderate behavioral alterations to severe ones.
Because prions are more robust than most germs, they are also deadly. Unlike bacteria or viruses, prions are not destroyed by cooking temperatures. There is no vaccination to prevent CJD or mechanism to build immunity to it because prions are immune system-blocking agents.
Diagnoses and Examinations
How is a diagnosis of Creutzfeldt-Jakob disease made?
Your healthcare professional may use a variety of techniques to diagnose CJD, such as:
Neurological and physical examinations: These entail your doctor searching for CJD symptoms and indicators and having you complete certain tasks that can be used to detect abnormalities in your brain’s functioning.
Imaging and diagnostic procedures: These tests have the ability to monitor brain activity and produce structural pictures of the brain.
Experiments in the lab: These tests look for indicators of CJD, particularly aberrant proteins and prions, in the blood or cerebrospinal fluid (the fluid that surrounds your brain and spinal cord). Testing brain tissue soon after a person passes away may also be part of this.





 – सांस्कृतिक धरोहरों को नया जीवन देने की पहल")





 has a rare disease that quickly deteriorates their brain. The symptoms of this disorder){kind=link}